



一個由密西根大學(University of Michigan)領導的研究團隊正在利用阿貢國家實驗室(Argonne National Laboratory)的超級電腦,開發大型基礎模型,以加速新型電池材料的發現。這些研究人員使用美國能源部的阿貢國家實驗室的 Aurora 和 Polaris 系統,開發能夠預測電池電解質和電極新材料的人工智慧(AI)模型。
長期以來,尋找更好的電池材料主要依賴試錯法。密西根大學的副教授Venkat Viswanathan表示:「在電池材料發現的歷史上,直覺一直是推動新發明的主要力量。」他指出,今天使用的大多數材料都是在1975年至1985年間發現的,至今仍主要依賴這些材料,僅進行小幅度的改進。
隨著人工智慧的進步及其所需的計算能力的提升,這一局面正在改變。Viswanathan和他的同事們正在開發AI基礎模型,以加速新電池材料的發現,這些材料可應用於個人電子產品和醫療設備等領域。
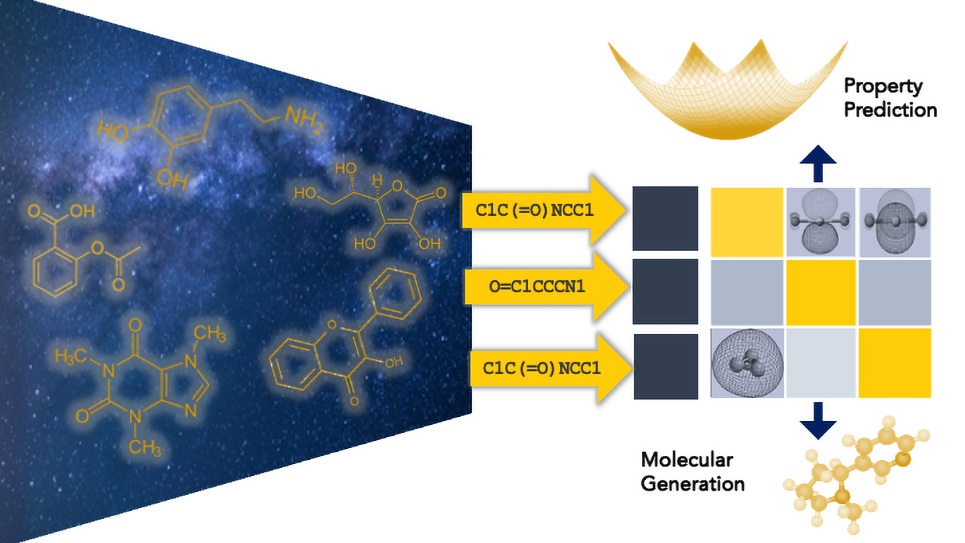
基礎模型是訓練於大量數據集上的大型AI系統,專門針對特定領域進行調整,與通用的大型語言模型(如ChatGPT)不同,這些科學基礎模型能夠生成更精確和可靠的預測。
▲ 密西根大學的研究人員正在利用阿貢國家實驗室的超級電腦,開發可加速分子設計與新電池材料發現的基礎模型。(Source:密西根大學)
該團隊的模型專注於辨識兩個關鍵電池組件的材料:電解質和電極。電解質負責傳遞電荷,而電極則儲存和釋放能量。為了設計出更強大、更持久且更安全的下一代電池,這兩方面的進步都是必需的。
潛在電池材料的化學空間規模龐大,科學家估計可能存在1,060種分子化合物。訓練於數十億已知分子的基礎模型能幫助研究人員更有效地探索這一空間,透過學習能預測新分子性質的模式,模型能夠鎖定高潛力候選者。
去年,Viswanathan的團隊使用Polaris超級電腦訓練了迄今為止最大的化學基礎模型之一,專注於設計電池電解質所需的小分子。為了教會模型理解分子結構,團隊使用SMILES系統,並開發了一種名為SMIRK的新工具,以提高模型處理這些結構的能力。
目前,研究人員正在利用阿貢國家領導級運算設施(ALCF)的新Aurora超級系統開發第二個基礎模型,專注於做為電池電極基礎的分子晶體。訓練完成後,基礎模型的預測結果將與實驗數據進行比較,以確保準確性,這對於建立對模型預測各種化學和物理性質的信心至關重要。
在開發基礎模型之前,Viswanathan的團隊曾為每個感興趣的性質開發較小的AI模型。訓練於Polaris的基礎模型不僅將這些能力統一在一個平台上,還超越了他們過去幾年創建的單一性質預測模型。
該團隊計劃將模型的能力擴展並在未來向更廣泛的研究社群開放,並與密西根大學的實驗室科學家合作,合成和測試AI模型辨識出的最有前景候選者。值得一提的是,密西根大學與美國能源部於2025年成立的「清潔能源儲存研究中心」專注於電池材料和技術創新,已獲7,500萬美元資助,與阿貢國家實驗室及其他12所大學合作,彰顯該研究的戰略重要性與資源支持。
(首圖來源:Argonne National Laboratory)





